################################################################################
################ ~ Prevalence of malaria infection full code ~ #################
################################################################################
### Step 1: Option 1 (Download Pre-Calculated Prevalence) ----------------------
#### Step 1.1.1: Install or load required packages -----------------------------
# install or load required packages
pacman::p_load(
here, # for handling relative file paths
haven, # for reading DHS .dta files and labelled data
dplyr, # for data wrangling
stringr, # for filtering U5MR rows using regex
tibble, # for rownames_to_column
ggplot2, # for visualizing U5MR maps
sf, # for working with shapefiles
rio, # for saving outputs in .csv and .rds formats
survey, # for complex survey analysis
cowplot, # for arranging plots
DHS.rates # for calculating under-five mortality rates
)
#' Check DHS Indicator List from API
#'
#' @param countryIds DHS country code(s), e.g., "EG"
#' @param indicatorIds specific indicator ID(s)
#' @param surveyIds survey ID(s)
#' @param surveyYear exact year
#' @param surveyYearStart start of year range
#' @param surveyYearEnd end of year range
#' @param surveyType DHS survey type (e.g., "DHS", "MIS")
#' @param surveyCharacteristicIds filter by survey characteristic ID
#' @param tagIds filter by tag ID
#' @param returnFields Fields to return
#' (default: IndicatorId, Label, Definition)
#' @param perPage max results per page (default = 500)
#' @param page specific page to return (default = 1)
#' @param f format (default = "json")
#'
#' @return a data.frame of indicators
#' @export
check_dhs_indicators <- function(
countryIds = NULL,
indicatorIds = NULL,
surveyIds = NULL,
surveyYear = NULL,
surveyYearStart = NULL,
surveyYearEnd = NULL,
surveyType = NULL,
surveyCharacteristicIds = NULL,
tagIds = NULL,
returnFields = c(
"IndicatorId", "Label", "Definition", "MeasurementType"
),
perPage = NULL,
page = NULL,
f = "json"
) {
# base URL
base_url <-
"https://api.dhsprogram.com/rest/dhs/indicators?"
# build query string
params <- list(
countryIds = countryIds,
indicatorIds = indicatorIds,
surveyIds = surveyIds,
surveyYear = surveyYear,
surveyYearStart = surveyYearStart,
surveyYearEnd = surveyYearEnd,
surveyType = surveyType,
surveyCharacteristicIds = surveyCharacteristicIds,
tagIds = tagIds,
returnFields = paste(returnFields, collapse = ","),
perPage = perPage,
page = page,
f = f
)
# drop NULLs and encode
query <- paste(
purrr::compact(params) |>
purrr::imap_chr(
~ paste0(
.y, "=", URLencode(as.character(.x), reserved = TRUE)
)
),
collapse = "&"
)
# full URL
full_url <- paste0(base_url, query)
# fetch with progress bar
response <- httr::GET(full_url, httr::progress())
jsonlite::fromJSON(httr::content(
response,
as = "text",
encoding = "UTF-8"
))$Data
}
#' Query DHS API Directly via URL Parameters
#'
#' builds and queries DHS API for indicator data using URL-based access
#' instead of rdhs package.
#'
#' @param countryIds Comma-separated DHS country code(s), e.g., "SL"
#' @param indicatorIds Comma-separated DHS indicator ID(s),
#' e.g., "CM_ECMR_C_U5M"
#' @param surveyIds optional comma-separated survey ID(s), e.g.,
#' "SL2016DHS"
#' @param surveyYear optional exact survey year, e.g., "2016"
#' @param surveyYearStart optional survey year range start
#' @param surveyYearEnd optional survey year range end
#' @param breakdown one of: "national", "subnational", "background",
#' "all"
#' @param f format to return (default is "json")
#'
#' @return a data.frame containing the `Data` portion of the API
#' response.
#' @export
download_dhs_indicators <- function(
countryIds,
indicatorIds,
surveyIds = NULL,
surveyYear = NULL,
surveyYearStart = NULL,
surveyYearEnd = NULL,
breakdown = "subnational",
f = "json"
) {
# base URL
base_url <-
"https://api.dhsprogram.com/rest/dhs/data?"
# assemble query string
query <- paste0(
"breakdown=",
breakdown,
"&indicatorIds=",
indicatorIds,
"&countryIds=",
countryIds,
if (!is.null(surveyIds))
paste0("&surveyIds=", surveyIds),
if (!is.null(surveyYear))
paste0("&surveyYear=", surveyYear),
if (!is.null(surveyYearStart))
paste0("&surveyYearStart=", surveyYearStart),
if (!is.null(surveyYearEnd))
paste0("&surveyYearEnd=", surveyYearEnd),
"&lang=en&f=",
f
)
full_url <- paste0(base_url, query)
cli::cli_alert_info("Downloading DHS data...")
response <- httr::GET(full_url, httr::progress())
if (httr::http_error(response)) {
stop("API request failed: ", httr::status_code(response))
}
content_raw <- httr::content(
response, as = "text", encoding = "UTF-8"
)
data <- jsonlite::fromJSON(content_raw)$Data
cli::cli_alert_success(
"Download complete: {nrow(data)} records retrieved."
)
return(data)
}
#### Step 1.1.2: Download the relevant indicators ------------------------------
# RDT-based prevalence
rdt_mean <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_RDT",
breakdown = "subnational"
) |>
dplyr::mutate(
dhs_indicator_id = "ML_PMAL_C_RDT",
indicator = "Malaria prevalence (RDT)"
) |>
dplyr::select(
dhs_indicator_id,
indicator,
region_label = CharacteristicLabel,
adm2_code = RegionId,
mean_prev = Value
)
rdt_low <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_RDL",
breakdown = "subnational"
) |>
dplyr::select(
region_label = CharacteristicLabel,
adm2_code = RegionId,
low_prev = Value
)
rdt_upp <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_RDU",
breakdown = "subnational"
) |>
dplyr::select(
region_label = CharacteristicLabel,
adm2_code = RegionId,
upp_prev = Value
)
# microscopy-based prevalence
mic_mean <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_MSY",
breakdown = "subnational"
) |>
dplyr::mutate(
dhs_indicator_id = "ML_PMAL_C_MSY",
indicator = "Malaria prevalence (Microscopy)"
) |>
dplyr::select(
dhs_indicator_id,
indicator,
region_label = CharacteristicLabel,
adm2_code = RegionId,
mean_prev = Value
)
mic_low <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_MSL",
breakdown = "subnational"
) |>
dplyr::select(
region_label = CharacteristicLabel,
adm2_code = RegionId,
low_prev = Value
)
mic_upp <- download_dhs_indicators(
countryIds = "SL",
surveyYear = 2016,
indicatorIds = "ML_PMAL_C_MSU",
breakdown = "subnational"
) |>
dplyr::select(
region_label = CharacteristicLabel,
adm2_code = RegionId,
upp_prev = Value
)
#### Step 1.1.3a: Join prevalence indicators when a dedicated shapefile IS available
# get the DHS adm2 shapefile
sle_dhs_shp2 <- sf::read_sf(
here::here(
"01_foundational/1a_administrative_boundaries",
"1ai_adm2",
"sdr_mis_subnational_boundaries_adm2.shp"
)
) |>
dplyr::select(
adm1 = OTHREGNA,
adm2 = DHSREGEN,
adm2_code = REG_ID
) |>
# make adm1 and adm2 to upper case
dplyr::mutate(
adm1 = toupper(adm1),
adm2 = toupper(adm2)
)
# join pfpr both indicators
pfpr_indicators <- dplyr::bind_rows(
pfpr_indicator_rdt,
pfpr_indicator_mic
)
# join adm2 dhs shapefile to data only keeping adm2 indicators
pfpr_indicators_final <- pfpr_indicators |>
dplyr::inner_join(sle_dhs_shp2, by = "adm2_code") |>
# select only relevant columns
dplyr::select(
dhs_indicator_id,
indicator,
adm1,
adm2,
mean_prev,
low_prev,
upp_prev,
mean_prev,
low_prev,
upp_prev,
) |>
sf::st_as_sf()
# check indicators
sf::st_drop_geometry(pfpr_indicators_final)
#### Step 1.1.3b: Join prevalence indicators when a dedicated shapefile is NOT available
# load the DHS `admin-2` shapefile
sle_dhs_shp2 <- sf::read_sf(
here::here(
"01_foundational/1a_administrative_boundaries",
"1ai_adm2",
"sdr_subnational_boundaries_adm2.shp"
)
) |>
dplyr::select(
adm1 = OTHREGNA,
adm2 = DHSREGEN,
adm2_code = REG_ID
) |>
# make adm1 and adm2 to upper case
dplyr::mutate(
adm1 = toupper(adm1),
adm2 = toupper(adm2)
)
# join rdt indicator with CIs
pfpr_indicator_rdt <- rdt_mean |>
dplyr::left_join(rdt_low, by = "region_label") |>
dplyr::left_join(rdt_upp, by = "region_label")
# join mic indicator with CIs
pfpr_indicator_mic <- mic_mean |>
dplyr::left_join(mic_low, by = "region_label") |>
dplyr::left_join(mic_upp, by = "region_label")
# combine RDT and microscopy indicators with CIs
pfpr_indicators <- dplyr::bind_rows(
pfpr_indicator_rdt,
pfpr_indicator_mic
) |>
# manually clean up admin names
dplyr::mutate(
region_label_updated = dplyr::case_when(
region_label == "..Kailahun" ~ "KAILAHUN",
region_label == "..Kenema" ~ "KENEMA",
region_label == "..Kono" ~ "KONO",
region_label == "..Koinadugu (before 2017)" ~ "KOINADUGU",
region_label == "..Tonkolili" ~ "TONKOLILI",
region_label == "..Kambia" ~ "KAMBIA",
region_label == "..Karene" ~ "KARENE",
region_label == "..Bombali (before 2017)" ~ "BOMBALI",
region_label == "..Port Loko (before 2017)" ~ "PORT LOKO",
region_label == "..Bo" ~ "BO",
region_label == "..Bonthe" ~ "BONTHE",
region_label == "..Moyamba" ~ "MOYAMBA",
region_label == "..Pujehun" ~ "PUJEHUN",
region_label == "..Western Rural" ~ "WESTERN AREA RURAL",
region_label == "..Western Urban" ~ "WESTERN AREA URBAN",
region_label == "Eastern" ~ "EASTERN",
region_label == "Northern (before 2017)" ~ "NORTHERN",
region_label == "Western" ~ "WESTERN",
region_label == "Southern" ~ "SOUTHERN",
TRUE ~ NA
)
) |>
dplyr::filter(!is.na(region_label_updated))
# join shapefile to indicator data
pfpr_indicators_final <-
pfpr_indicators |>
dplyr::inner_join(sle_dhs_shp2, by = c("region_label_updated" = "adm2")) |>
dplyr::select(
dhs_indicator_id,
indicator,
adm1,
adm2 = region_label_updated,
mean_prev,
low_prev,
upp_prev,
geometry
) |>
sf::st_as_sf()
# preview the joined data (without geometry)
sf::st_drop_geometry(pfpr_indicators_final)
### Step 1: Option 2 (Calculate Prevalence from Raw DHS Data) ------------------
#### Step 1.2.1: Load the relevant DHS data ------------------------------------
# import the KR (children's) data
sle_dhs_pr <- readRDS(
here::here("1.6_health_systems/1.6a_dhs/raw/SLPR73FL.rds")
) |>
# make location column
dplyr::mutate(
adm1 = haven::as_factor(hv024) |>
toupper(),
adm2 = haven::as_factor(shdist) |>
toupper()
)
#### Step 1.2.2: Extract individual records ------------------------------------
# extract pfpr from individual records
sle_dhs_pr2 <- sle_dhs_pr |>
dplyr::mutate(
# tested for malaria via RDT
tested_rdt = dplyr::case_when(
# child was present last night (hv103 == 1),
# has a mother in the household (hv042 == 1),
# is between 6–59 months (hc1),
# and has a valid RDT result (0 = negative, 1 = positive)
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml35 %in% c(0, 1) ~ 1,
# child meets criteria but no valid RDT result recorded
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 ~ 0,
TRUE ~ NA_real_ # all others set to missing
),
# microscopy testing status
tested_mic = dplyr::case_when(
# child was present last night (hv103 == 1),
# has a mother in the household (hv042 == 1),
# is aged 6–59 months (hc1),
# and has a microscopy result: 0 = negative,
# 1 = positive, 6 = invalid (still considered tested)
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml32 %in% c(0, 1, 6) ~
1,
# child is eligible but no valid microscopy result available
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 ~ 0,
TRUE ~ NA_real_ # all others set to missing
),
# RDT positive
rdt_pos = dplyr::case_when(
# positive RDT result (hml35 == 1) for eligible child
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml35 == 1 ~ 1,
# negative RDT result (hml35 == 0) for eligible child
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml35 == 0 ~ 0,
TRUE ~ NA_real_ # all others set to missing
),
# microscopy positive
mic_pos = dplyr::case_when(
# positive microscopy result (hml32 == 1) for eligible child
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml32 == 1 ~ 1,
# negative or invalid microscopy result (0 or 6)
hv103 == 1 & hv042 == 1 & hc1 >= 6 & hc1 <= 59 &
hml32 %in% c(0, 6) ~ 0,
TRUE ~ NA_real_ # all others set to missing
),
survey_weight = hv005 / 1000000
) |>
dplyr::select(
cluster_id = hv021,
stratum_id = hv022,
survey_weight,
adm1,
adm2,
tested_rdt,
tested_mic,
rdt_pos,
mic_pos
)
#### Step 1.2.3: Calculate malaria prevalence (RDT and Microscopy) at district level
# RDT design
des_rdt <- survey::svydesign(
ids = ~cluster_id,
strata = ~stratum_id,
weights = ~survey_weight,
data = dplyr::filter(sle_dhs_pr2, tested_rdt == 1),
nest = TRUE
)
# microscopy design
des_mic <- survey::svydesign(
ids = ~cluster_id,
strata = ~stratum_id,
weights = ~survey_weight,
data = dplyr::filter(sle_dhs_pr2, tested_mic == 1),
nest = TRUE
)
# RDT prevalence by adm2
prev_rdt <- survey::svyby(
~rdt_pos,
~ adm1 + adm2,
design = des_rdt,
FUN = survey::svymean,
vartype = c("ci"), # add CI
keep.names = F
) |>
dplyr::mutate(
pct = round(rdt_pos * 100, 1),
ci_low = round(ci_l * 100, 1),
ci_upp = round(ci_u * 100, 1)
) |>
dplyr::arrange(adm1) |>
dplyr::mutate(indicator = "Malaria prevalence (RDT)") |>
dplyr::select(adm2, indicator, pct, ci_low, ci_upp)
# microscopy prevalence by adm2
prev_mic <- survey::svyby(
~mic_pos,
~ adm1 + adm2,
design = des_mic,
FUN = survey::svymean,
vartype = c("ci"), # add CI
keep.names = F
) |>
dplyr::mutate(
pct = round(mic_pos * 100, 1),
ci_low = round(ci_l * 100, 1),
ci_upp = round(ci_u * 100, 1)
) |>
dplyr::arrange(adm1) |>
dplyr::mutate(indicator = "Malaria prevalence (Microscopy)") |>
dplyr::select(adm2, indicator, pct, ci_low, ci_upp)
# bind both together
prev_final <- dplyr::bind_rows(prev_rdt, prev_mic)
#### Step 1.2.4: Join prevalence indicators to a shapefile ---------------------
# load the DHS `admin-2` shapefile
sle_dhs_shp2 <- sf::read_sf(
here::here(
"01_foundational/1a_administrative_boundaries",
"1ai_adm2",
"sdr_subnational_boundaries_adm2.shp"
)
) |>
dplyr::select(
adm1 = OTHREGNA,
adm2 = DHSREGEN,
adm2_code = REG_ID
) |>
# make adm1 and adm2 to upper case
dplyr::mutate(
adm1 = toupper(adm1),
adm2 = toupper(adm2)
)
# bind both together
prev_final <- prev_final |>
# clean up adm2 names to match shapefile
dplyr::mutate(
adm2 = dplyr::case_when(
adm2 == "WEST AREA RURAL" ~ "WESTERN AREA RURAL",
adm2 == "WEST AREA URBAN" ~ "WESTERN AREA URBAN",
TRUE ~ adm2
)
) |>
# join to shapefile
dplyr::left_join(sle_dhs_shp2, by = c("adm2")) |>
sf::st_as_sf()
sf::st_drop_geometry(prev_final)
### Step 2: Visualize Subnational Indicator Data -------------------------------
# categorize prevalence percentages
prev_final <- prev_final |>
dplyr::mutate(
prev_cat = cut(
pct,
breaks = c(0, 10, 20, 30, 40, 50, 60, 70, 80, 90, 100),
labels = c(
"<10",
"10 to <20",
"20 to <30",
"30 to <40",
"40 to <50",
"50 to <60",
"60 to <70",
"70 to <80",
"80 to <90",
"90 to 100"
),
include.lowest = TRUE,
right = FALSE
)
)
# visualize RDT-based prevalence
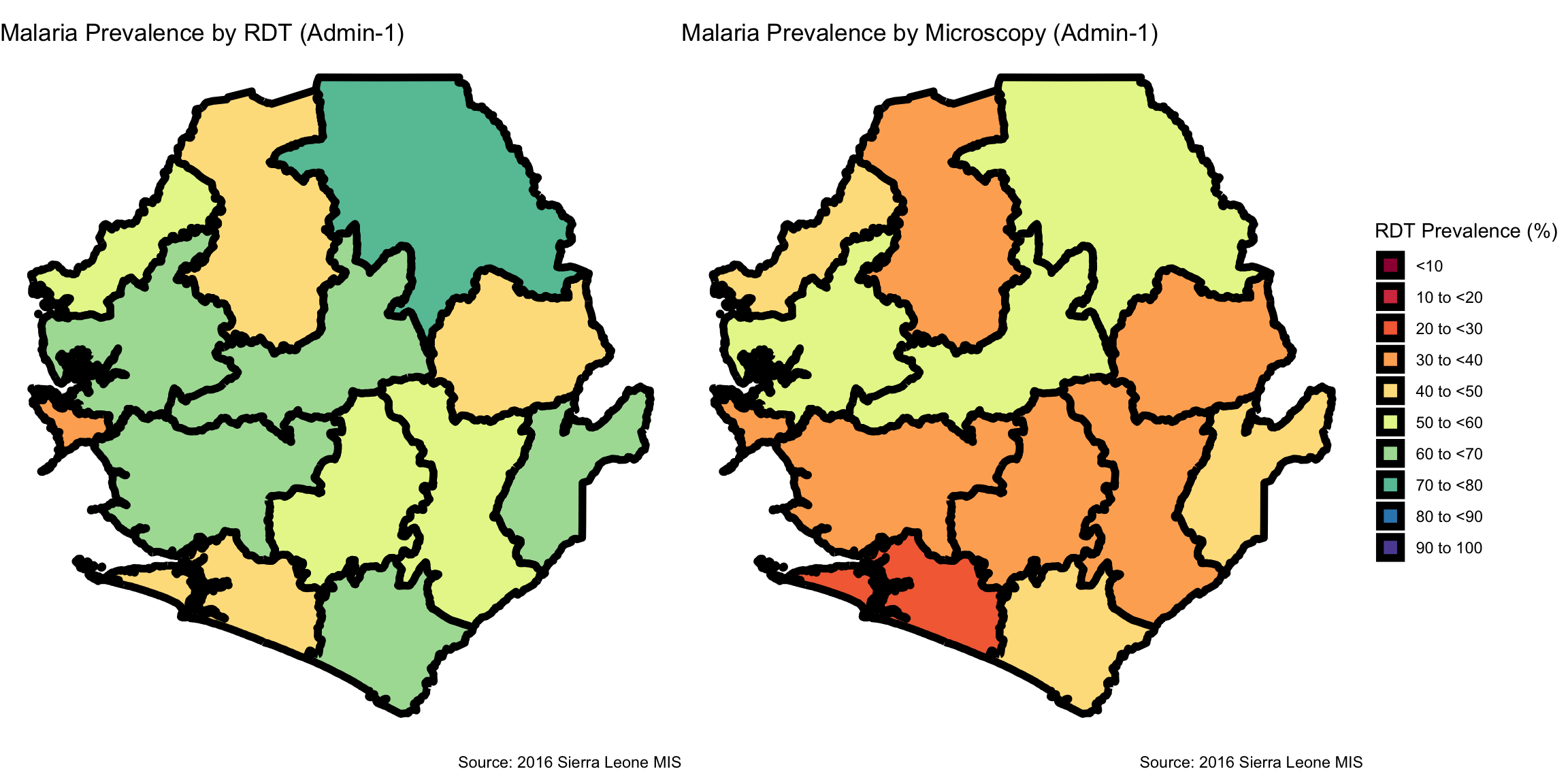
map_rdt <- prev_final |>
dplyr::filter(indicator == "Malaria prevalence (RDT)") |>
ggplot2::ggplot() +
ggplot2::geom_sf(
ggplot2::aes(fill = prev_cat),
color = "black",
size = 2,
show.legend = TRUE
) +
ggplot2::scale_fill_manual(
name = "RDT Prevalence (%)",
values = c(
"<10" = "#9E0142",
"10 to <20" = "#D53E4F",
"20 to <30" = "#F46D43",
"30 to <40" = "#FDAE61",
"40 to <50" = "#FEE08B",
"50 to <60" = "#E6F598",
"60 to <70" = "#ABDDA4",
"70 to <80" = "#66C2A5",
"80 to <90" = "#3288BD",
"90 to 100" = "#5E4FA2"
),
drop = FALSE
) +
ggplot2::labs(
title = "Malaria Prevalence by RDT (Admin-1)",
caption = "Source: 2016 Sierra Leone MIS"
) +
ggplot2::theme_void()
# microscopy map
map_mic <- prev_final |>
dplyr::filter(indicator != "Malaria prevalence (RDT)") |>
ggplot2::ggplot() +
ggplot2::geom_sf(
ggplot2::aes(fill = prev_cat),
color = "black",
size = 2,
show.legend = TRUE
) +
ggplot2::scale_fill_manual(
name = "Microscopy Prevalence (%)",
values = c(
"<10" = "#9E0142",
"10 to <20" = "#D53E4F",
"20 to <30" = "#F46D43",
"30 to <40" = "#FDAE61",
"40 to <50" = "#FEE08B",
"50 to <60" = "#E6F598",
"60 to <70" = "#ABDDA4",
"70 to <80" = "#66C2A5",
"80 to <90" = "#3288BD",
"90 to 100" = "#5E4FA2"
),
drop = FALSE
) +
ggplot2::labs(
title = "Malaria Prevalence by Microscopy (Admin-1)",
caption = "Source: 2016 Sierra Leone MIS"
) +
ggplot2::theme_void()
# display maps side-by-side with shared legend
cowplot::plot_grid(
map_rdt + ggplot2::theme(legend.position = "none"),
map_mic + ggplot2::theme(legend.position = "none"),
cowplot::get_legend(map_rdt),
ncol = 3,
rel_widths = c(1, 1, 0.3)
)
# save RDT map
ggplot2::ggsave(
plot = map_rdt,
filename = here::here(
"03_output/3a_figures/malaria_prev_rdt_adm2.png"
),
width = 12,
height = 9,
dpi = 300
)
# save microscopy map
ggplot2::ggsave(
plot = map_mic,
filename = here::here(
"03_output/3a_figures/malaria_prev_mic_adm2.png"
),
width = 12,
height = 9,
dpi = 300
)
### Step 3: Save Processed Malaria Prevalence Indicators -----------------------
# define save directory
save_path <- here::here("1.6_health_systems/1.6a_dhs")
# save final joined malaria prevalence data
rio::export(
prev_final |> sf::st_drop_geometry(),
here::here(
save_path, "processed", "final_malaria_prev_sle_adm2.csv"
)
)
# save final joined malaria prevalence data
rio::export(
prev_final,
here::here(
save_path, "processed", "final_malaria_prev_sle_adm2.rds"
)
)